Related Subjects:
|Osteoporosis
|Malabsorption
|Bronchiectasis
|Pseudomonas infection (Pseudomonas aeruginosa)
|Autosomal Recessive
|Genetic Mutations
|Clustered Regularly Interspaced Short Palindromic Repeats CRISPR
Disease presents without warning and some forms milder than others. A sweat chloride concentration of 80 mmol/l is diagnostic. A person with CF cannot contract cholera, because the toxin cannot open the chloride channels in the small intestine. With improved supportive care, the median survival in the UK is now more than 50 years.
Curbed flows of chloride
Lung’s massed mucus, thick and dried
Bad bugs stick and thrive
Body’s small tubes have it worst
Baby gut often blocked first
@DrCindyCooper
About
- Inherited Defect of the Cystic fibrosis transmembrane conductance regulator (CFTR)
- Leads to the production of thick viscid mucus and secondary infection
Aetiology
- CF is inherited as an AR and there are at least 800 gene mutations
- Mutation in cystic fibrosis transmembrane conductance regulator (CFTR) gene on 7q31.2.
- Genotype is a poor guide to disease severity even within families possibly due to unknown modifier genes
- Over 70% are due to the delta F508 mutation on chromosome 7q which codes for the CFTR
- Incidence in the UK is 1 in 2500 and life expectancy for teenagers is 40-50 years old
Gene Mutations and effects
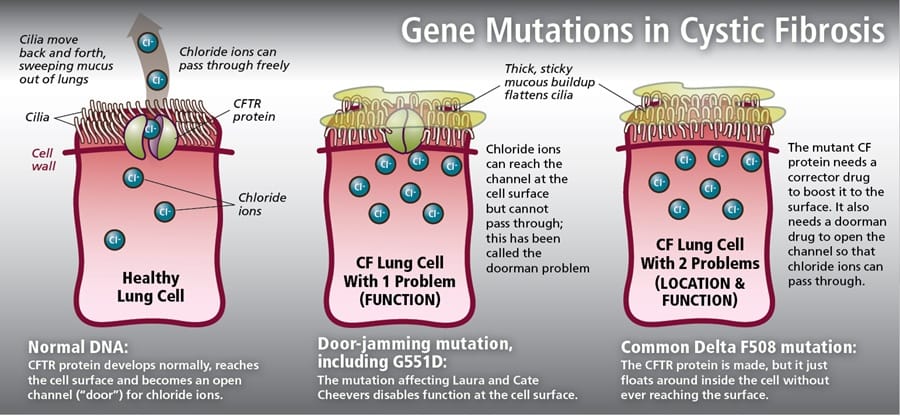
Diagnosis
- Positive test results in people with no symptoms, for example infant screening (blood spot immunoreactive trypsin test) followed by sweat and gene tests for confirmation or
- clinical manifestations, supported by sweat or gene test results for confirmation or
- Clinical manifestations alone, in the rare case of people with symptoms who have normal sweat or gene test results.
Clinical Gastrointestinal
- Failure to thrive and Malabsorption
- Meconium ileus - intestinal obstruction at birth
- Meconium ileus equivalent later
- Gallstone ileus and gallstone related disease
- Pancreatic insufficiency - steatorrhoea, weight loss
- Increased incidence of cholecystitis and gallstones
- Fatty liver and focal biliary cirrhosis
- Secondary Diabetes develops in 25% of patients
- Rectal prolapse
Pulmonary
- Normal lungs become progressively damaged
- Recurrent chest symptoms - infections, bronchiectasis
- Pneumothorax - seen mainly in adults
- Massive haemoptysis, Bronchitis, bronchiolitis, Bronchiectasis
- Eventual Respiratory failure, Rhinitis and nasal polyps
- Overtime increasing Pseudomonas infection
- Respiratory infections: Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa
Others
- Finger clubbing and Nasal polyps and sinusitis
- Circulating immune complexes and amyloidosis
- Male infertility - Failure to develop epididymis and vas deferens
- Osteoporosis and rickets
- Infant's sweat tastes salty - may be noted by parent
Carriers of CF enjoy the mixed blessing of a balanced polymorphism. They do not have enough abnormal chloride channels to cause the laboured breathing and clogged pancreas of cystic fibrosis, but they do have enough of a defect to prevent cholera from taking hold. During the devastating cholera epidemics that have peppered history, individuals carrying mutant CF alleles had a selective advantage, and they disproportionately transmitted those alleles to future generations. However, because CF arose in Western Europe and cholera in Africa, perhaps an initial increase in CF heterozygosity was a response to a different diarrheal infection
Investigations
- Immunoreactive trypsinogen is elevated in new-borns. This will need a sweat test if positive
- Sweat test: CF is diagnosed by clinical suspicion followed by a sweat test using Pilocarpine iontophoresis. If the sweat [Cl] or [Na] > 60 mmol/l then the test is considered positive. Two reliable positive sweat tests with a chloride concentration > 60 mmol/l is considered to be diagnostic
- DNA analysis may be done but with a large number of mutations is not needed
- Pancreatic insufficiency - faecal elastase, ? faecal fats, ? glucose with secondary diabetes
- USS testes may show an absent vas deferens and epididymis
- Establish baseline respiratory function testing
Summary of testing for CF
| Diagnostic Test |
Description |
Diagnostic Criteria |
| Sweat Chloride Test |
Measures the concentration of chloride in sweat. It is the primary test used to diagnose CF. |
- ≥ 60 mmol/L: Positive for CF (Diagnostic)
- 30-59 mmol/L: Intermediate (Possible CF, further testing required)
- < 30 mmol/L: CF unlikely
|
| Genetic Testing |
Identification of mutations in the CFTR gene. |
- Presence of two pathogenic mutations in the CFTR gene confirms CF diagnosis.
- Genetic testing is also used for prenatal screening and carrier testing.
|
| Nasal Potential Difference (NPD) Test |
Measures the voltage across the nasal epithelium to assess ion transport defects associated with CF. |
- Abnormal NPD readings support CF diagnosis, particularly in atypical or unclear cases.
|
| Newborn Screening (Immunoreactive Trypsinogen - IRT) |
Measures the level of immunoreactive trypsinogen in the blood. Elevated levels may indicate CF. |
- High IRT levels prompt further testing, such as a sweat chloride test or genetic testing.
|
| Clinical Presentation |
Signs and symptoms suggestive of CF, including chronic respiratory issues, failure to thrive, and gastrointestinal symptoms. |
- Presence of typical CF symptoms along with positive diagnostic tests (e.g., sweat chloride test) confirms CF diagnosis.
|
Complications
- Being underweight
- Meconium ileus (affects 1 in 7 newborn babies)
- Fat-soluble vitamin deficiencies (including vitamins A, D, E and K)
- Distal intestinal obstruction syndrome
- Muscle pains and arthralgia
- Male infertility caused by obstructive azoospermia (almost all males with cystic fibrosis are infertile)
- Reduced female fertility
- Upper airway complications, including nasal polyps and sinusitis (prevalence increases with age)
- Chronic liver disease (the prevalence increases with age until early adulthood)
- Urinary stress incontinence
- Cystic-fibrosis-related diabetes (uncommon in children under 10 years, but the prevalence increases with age and it affects up to 1 in 2 adults)
- Reduced bone mineral density (including osteoporosis).
- Cystic-fibrosis-related arthritis
- Delayed puberty (associated with severe cystic fibrosis)
- Renal calculi (incidence increases with age and 1 in 20 adults are affected).
Management
| Management Area |
Interventions |
Comments |
| Airway Clearance |
Chest physiotherapy, postural drainage, oscillating positive expiratory pressure (PEP) devices, high-frequency chest wall oscillation (HFCWO). |
Regular airway clearance is critical to reduce mucus build-up and prevent infections. |
| Dornase alpha |
Dornase alfa is a genetically engineered version of a naturally occurring human enzyme which cleaves extracellular deoxyribonucleic acid (DNA). |
Management of cystic fibrosis patients with a forced vital capacity (FVC) of greater than 40% of predicted to improve pulmonary function. By inhalation of nebulised solution
Adult 2500 units once daily, administered by jet nebuliser, patients over 21 years may benefit from twice daily dosage. |
| Antibiotic Therapy |
Oral, inhaled, or IV tailored to sputum culture results, long-term suppressive therapy (e.g., azithromycin). |
Inhaled antibiotics e.g. nebulised tobramycin used for chronic Pseudomonas aeruginosa infections in older children. Rotate antibiotics to minimize resistance. Prophylactic azithromycin may be used for Pseudomonas. |
| Steroids |
For acute exacerbation of superimposed aspergillus infections |
Short courses. Watch for diabetes. |
| Nutritional Support |
Pancreatic enzyme replacement therapy e.g. Creon capsules / PERT, fat-soluble vitamin supplementation, high-calorie diet, laxatives. |
Most need PERT, nutritional support to prevent malnutrition. |
| Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators |
Ivacaftor, Lumacaftor/Ivacaftor, Tezacaftor/Ivacaftor, Elexacaftor/Tezacaftor/Ivacaftor. |
CFTR modulators target the underlying genetic defect in CF; prescribed based on the patient's specific CFTR mutations. |
| Management of Complications |
Management of CF-related diabetes (CFRD), bone disease (osteoporosis), liver disease, and gastrointestinal complications. |
Regular screening and proactive management of complications are essential for improving long-term outcomes. |
| Psychosocial Support |
Counselling, mental health support, social work services, patient education. |
Addressing the psychological and social aspects of CF is important for overall quality of life. |
| Male Infertility |
Refer for specialist help and genetic counselling. |
Various male fertility measures can be used to assist conception such as testicular sperm extraction (TEST), and intracytoplasmic sperm injection. |
| Lung Transplantation |
Consideration for patients with advanced lung disease unresponsive to medical management. |
Lung transplantation can significantly improve survival in end-stage CF, but careful selection and timing are crucial. |
| Osteoporosis |
Monitor, consider DEXA. |
Add Calcium/Vitamin D, Bisphosphonates. |
| Monitoring and Follow-up |
Regular spirometry, sputum cultures, nutritional assessments, liver function tests, glucose tolerance tests as a risk of diabetes with pancreatic failure. |
Multidisciplinary team approach with regular follow-up in specialized CF centres is recommended. |
References