Makindo Medical Notes.com |
|
|---|---|
| Download all this content in the Apps now Android App and Apple iPhone/Pad App | |
| MEDICAL DISCLAIMER:The contents are under continuing development and improvements and despite all efforts may contain errors of omission or fact. This is not to be used for the assessment, diagnosis or management of patients. It should not be regarded as medical advice by healthcare workers or laypeople. It is for educational purposes only. Please adhere to your local protocols. Use the BNF for drug information. If you are unwell please seek urgent healthcare advice. If you do not accept this then please do not use the website. Makindo Ltd | |
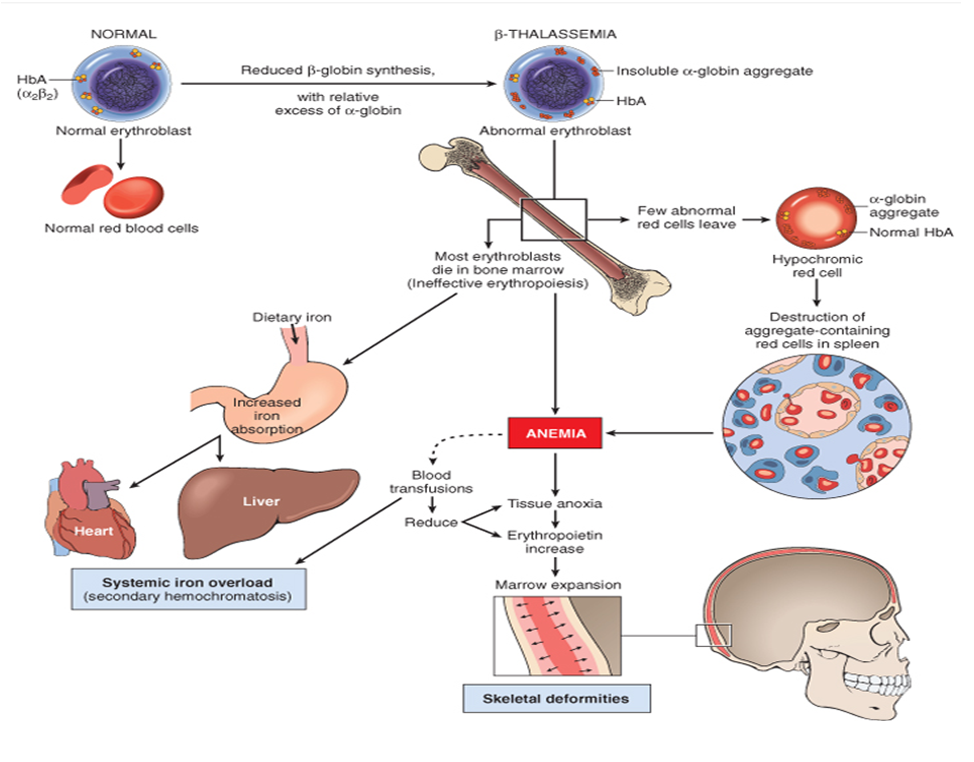
Beta Thalassaemia
-
| About | Anaesthetics and Critical Care | Anatomy | Biochemistry | Cardiology | Clinical Cases | CompSci | Crib | Dermatology | Differentials | Drugs | ENT | Electrocardiogram | Embryology | Emergency Medicine | Endocrinology | Ethics | Foundation Doctors | Gastroenterology | General Information | General Practice | Genetics | Geriatric Medicine | Guidelines | Haematology | Hepatology | Immunology | Infectious Diseases | Infographic | Investigations | Lists | Microbiology | Miscellaneous | Nephrology | Neuroanatomy | Neurology | Nutrition | OSCE | Obstetrics Gynaecology | Oncology | Ophthalmology | Oral Medicine and Dentistry | Paediatrics | Palliative | Pathology | Pharmacology | Physiology | Procedures | Psychiatry | Radiology | Respiratory | Resuscitation | Rheumatology | Statistics and Research | Stroke | Surgery | Toxicology | Trauma and Orthopaedics | Twitter | Urology
Related Subjects:
|Alpha Thalassaemia
|Beta Thalassaemia
|Anaemia of Chronic Disease
|Aplastic anaemia(AML)
|Autoimmune Haemolytic anaemia (AIHA)
Trait is common with a hypochromic microcytic anaemia with target cells and increased HbA2 on electrophoresis
About
Epidemiology
Aetiology
Classification
Clinical
Investigations
Management
| PLEASE NOTE LEGAL ADVICE: The contents are under continuing development and improvements and may contain errors of omission or fact. Feedback vital and always welcome at drokane at gmail.com. This is not to be used for the assessment, diagnosis or management of patients. It should not be regarded as medical advice. It is only for educational purposes. Please adhere to your local protocols. If you are unwell please seek healthcare advice from your doctor. This does not replace senior or specialist advice. If you do not accept this then please do not use the website. If you need medical advice, please consult a doctor or other appropriate medical professional. If you are a medical professional and you need advice then speak to your senior or colleagues. |